
You are here
Science Resources: DNA Technologies
DNA Sequencing Technologies, How They Differ, and Why It Matters
Sequencing methods have evolved rapidly over the past several years, and they are continually being developed. Today, there are three “generations” of sequencing technologies. These technologies differ in their methods for reading DNA and their ability to carry out multiple parallel sequencing reactions. Consequently, they vary in the speed of sequencing (throughput), the length of DNA they sequence (read length), and the ability to correctly determine the true DNA sequence (error rate). All three generations of sequencing technologies are still used today, and, sometimes, multiple technologies are used at the same time. Deciding which sequencing technology to use depends on a number of factors, including a laboratory’s familiarity and access to a technology along with requirements for speed, accuracy, and mobility.
Sanger Sequencing: The First Generation and Gold Standard for Sequencing
Sanger sequencing is the foundational method for sequencing DNA.[1] It was the only method of DNA sequencing available until second and third generation technologies, collectively known as next generation sequencing (NGS), emerged in the 1990s. Sanger sequencing has become less widely used today due to its low throughput (a result of low speed and limited parallelization), high price, and shorter read length (~1,000 base pairs (bp), Table 1). [2] It continues to be used as a critical validation method for targeted sequencing when confirming medical genetic tests or when designing new sequencing technologies.
Table 1. Comparison of Different Generations of Sequencing Technologies[3]
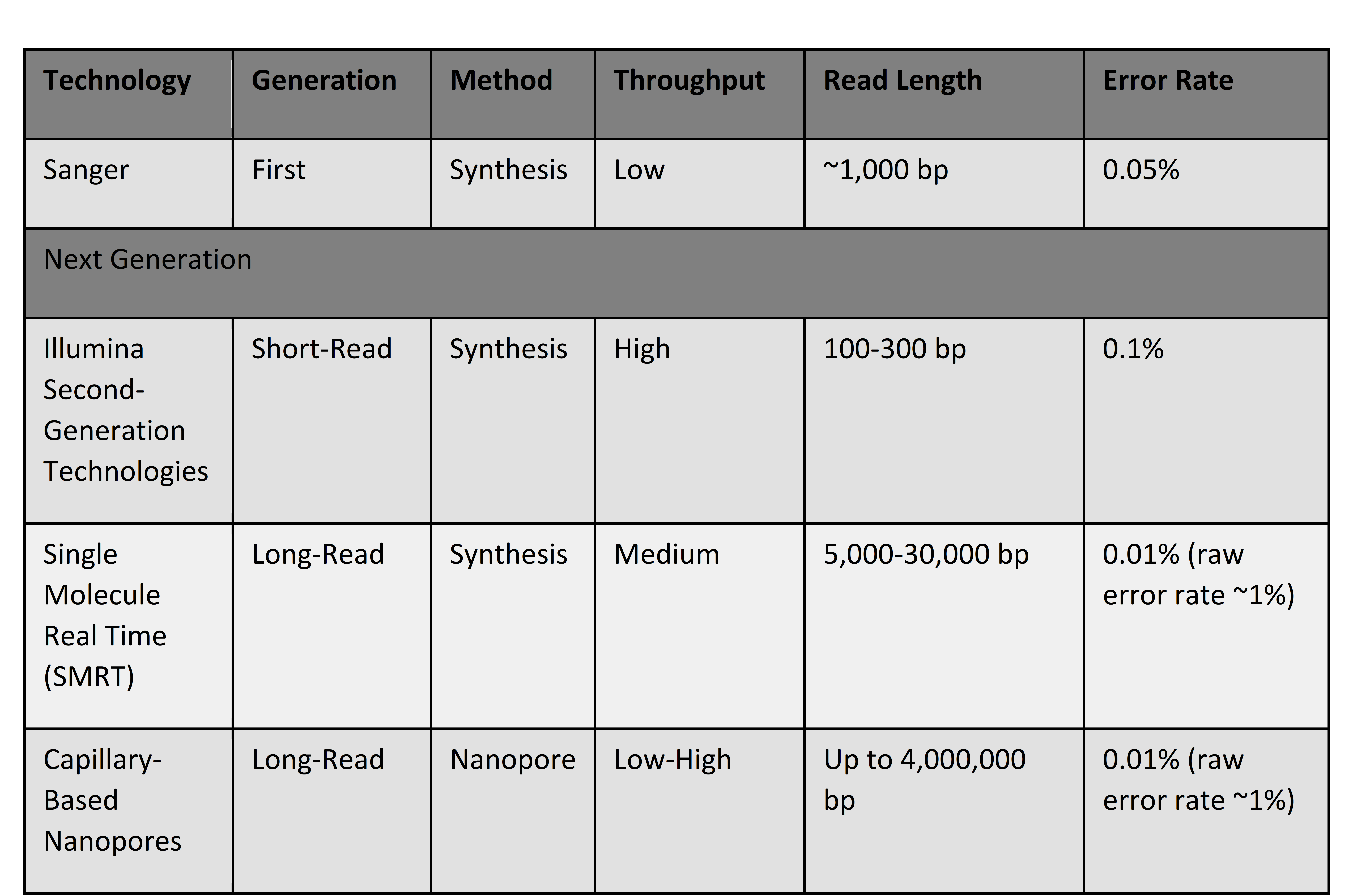
Note: Error rates for next-generation technologies can be reduced with additional methods, like duplex sequencing. Error rates for SMRT and nanopore technologies are continually advancing. Error rates described above are circa 2023.
Litigation that involves the reliability or accuracy of a medical genetic test result may include supplemental evidence from Sanger sequencing to confirm the initial test result. Any litigation related to patents of new sequencing technologies or claims about their accuracy and reliability may include evidence from Sanger sequencing to validate the new technology as well.
Sanger sequencing also provides a useful structure for describing sequencing technologies. The sequencing process involves three sequential steps (Fig. 7):
- Build/synthesize DNA segments complementary to the DNA region of interest.
The DNA region targeted for sequencing is used as a template to synthesize complementary DNA segments (Fig. 7A). During the synthesis of identical segments, chemically modified nucleotide bases are used in addition to regular nucleotide bases. The modified bases are fluorescently labeled terminating bases, meaning they are fluorescently labeled according to their identity (i.e., A, C, T, G) in order to be identified by a scanning computer, and they prevent further bases from being attached after they are incorporated into the new DNA segment.
During the synthesis of new DNA segments, the fluorescently labeled terminating bases are randomly incorporated. This produces multiple DNA segments representing varying levels of completeness of the original target DNA region (Fig. 7A). Each of these segments ends with a fluorescently labeled base identifying the last nucleotide to be incorporated (Fig. 7A).
- Separate synthesized DNA fragments by size.
Newly synthesized DNA segments of different lengths are size-separated using a capillary gel matrix and an electrical current (Fig. 7B). Smaller segments move more quickly through the gel than larger segments; a DNA fragment with the first base labeled moves to the bottom of the gel, and a segment with the final base labeled stays toward the top of the gel.
- Detect the DNA region’s sequence using the nucleotides’ fluorescence.
The size-separated DNA segments are read by a laser-detection system that records the color of the fluorescence attached to the segment (Fig. 7B). The fluorescence is read by a computer to determine the identity of each terminal base and its position within the overall DNA sequence (Fig. 7C). In this way, the sequentially terminated segments are pieced together to reconstruct the complete sequence of the target DNA region.

Figure 7. Schematic of the steps in Sanger sequencing. A) The target DNA region is used as a template for synthesizing new, identical DNA segments that incorporate fluorescently labeled terminating nucleotides, producing an array of different-sized DNA segments with the terminal base identifiable. B) The labeled DNA segments are size-separated in a gel matrix. C) The target DNA region’s sequence is reconstructed by detecting the terminal nucleotide for each DNA segment, from smallest to largest, using a scanning laser and computer detector.
Short-Read Sequencing: The Second Generation of Faster and Cheaper Sequencing
Some of the common sequencing technologies today are part of the second-generation sequencing technologies.[4] They are used routinely in medical genetic testing, research, and commercial/direct-to-consumer genetic services.
The basic method for these technologies is to sequence millions of short fragments of DNA (100–300 bp) (Table 1) simultaneously before combining the segments into larger sequences, like a whole genome, using computer software. Compared to Sanger sequencing, next-generation sequencing can produce a large amount of DNA sequence quickly (high throughput) and cheaply. The largest brand for second-generation sequencers is Illumina, whose sequencing technologies use a method known as sequencing-by-synthesis (SBS). This method involves four steps:
- Fragment the DNA into short segments and attach adapters.
DNA from a sample is fragmented into short segments (i.e., short-reads) (Fig. 8A), and small DNA sequences known as adapters are attached to the ends to facilitate collecting the DNA fragments. This step is referred to as library preparation, and the small pieces of DNA are the DNA library. Molecular probes can be used to pull out specific regions of DNA if a targeted region of the genome is being sequenced.
- Affix DNA segments to silica chip and duplicate segments.
The DNA library fragments are split into single-stranded DNA molecules and diffusely bound to a silica chip using the adapters (Fig. 8B). These fragments are then copied to make dense clusters of identical fragments (bridge amplification), which improves the sequencer’s signal detection in later steps.
- Determine the sequence of DNA segments by sequentially adding fluorescent nucleotides.
Fluorescently labeled nucleotide bases are repeatedly washed across the surface of the plate and bind to the DNA strands affixed to the surface (Fig. 8C). The nucleotide bases bind to the amplified DNA-library fragments affixed to the plate, building new, complementary DNA fragments that are localized in place.
As each base is incorporated, its fluorescence is recorded by a computer scanning the plate (Fig. 8C). Unlike in Sanger sequencing, these fluorescent bases do not permanently block the incorporation of subsequent bases. New DNA strands are thereby synthesized by repeatedly adding new, labeled bases, and their sequence is determined simultaneously—hence “sequencing-by-synthesis.” Across the chip, the sequences for millions of different DNA strands are determined in parallel.
- Use computer software to combine the sequences for short fragments into a single larger sequence.
Computer software assembles the sequences for the small DNA fragments into a larger sequence, for example a whole genome, by identifying overlapping regions (Fig. 8D). If a reference genome exists, like the human reference genome, it can be used as a guide to align and map short reads to their correct locations. Reference genomes vary in quality and do not exist for all organisms.
The entire sequencing process is performed in a fully automated sequencer. Because the sequencer needs to house reagents, robotics, camera, and computer, second-generation sequencers tend to be large, ranging from the size of a desktop computer to the size of a small refrigerator. Second-generation sequencers often have a designated location in the lab and are not mobile.
.png)
Figure 8. Schematic of short-read sequencing. A) Fragment the DNA into short segments and attach adapters. B) Affix DNA segments to a silica chip and duplicate segments. C) Determine the sequence of DNA segments by sequentially adding fluorescent nucleotides. D) Use computer software to combine the sequences for short fragments into a single larger sequence.
Long-Read Sequencing: A Third Generation of Technologies Expands the Scope of Sequencing
A new generation of sequencing technologies, third-generation sequencing (TGS) technologies, are now available and have greatly increased in prominence.[5] These methods distinguish themselves by being able to sequence much longer segments of DNA (10,0000–1,000,000 bp). In addition to detecting single-base mutations like second-generation technologies, these new sequencing methods are better able to detect structural variation in the DNA, including duplications and rearrangements of the DNA sequence.
Long-read sequencing methods tend to have higher initial error rates than short-read sequencing, reaching close to 15% (Table 1).[6] The high error rate compared to second-generation sequencing can be overcome with computational and molecular adaptations, including circularizing the DNA fragment to enable repeated resequencing and generate a consensus for the true sequence. These adaptations can bring the error rate closer to 0.01% (Table 1).[7] Duplex sequencing can also be used to reduce initial error rates, especially when detecting single nucleotide variants. In duplex sequencing, each strand of DNA is sequenced independently, and then a "consensus sequence" is assembled from the two strands. This method reduces initial error rates up to 10-million-fold below traditional next generation methods.[8]
There are two major platforms for long-read sequencing, one developed by Pacific Biosciences and the other developed by Oxford Nanopore Technologies.[9] Each uses a different method for sequencing long segments of DNA.
Pacific Biosciences' HiFi Sequencing uses single molecule, real-time (SMRT) sequencing (Fig. 9), which is similar in ways to sequencing-by-synthesis.[10] In SMRT sequencing, long segments of DNA are diluted into microscopic wells that cover the surface of a glass plate (Fig. 9A). Each well holds a single DNA molecule and a single DNA polymerase, which reads the DNA segment and synthesizes a new, complementary DNA strand using fluorescently labeled nucleotides (Fig. 9B). As nucleotides are strung together, they emit characteristic pulses of light that are recorded by a computer (Fig. 9B). The computer monitors the thousands of strands being synthesized simultaneously in all the wells.
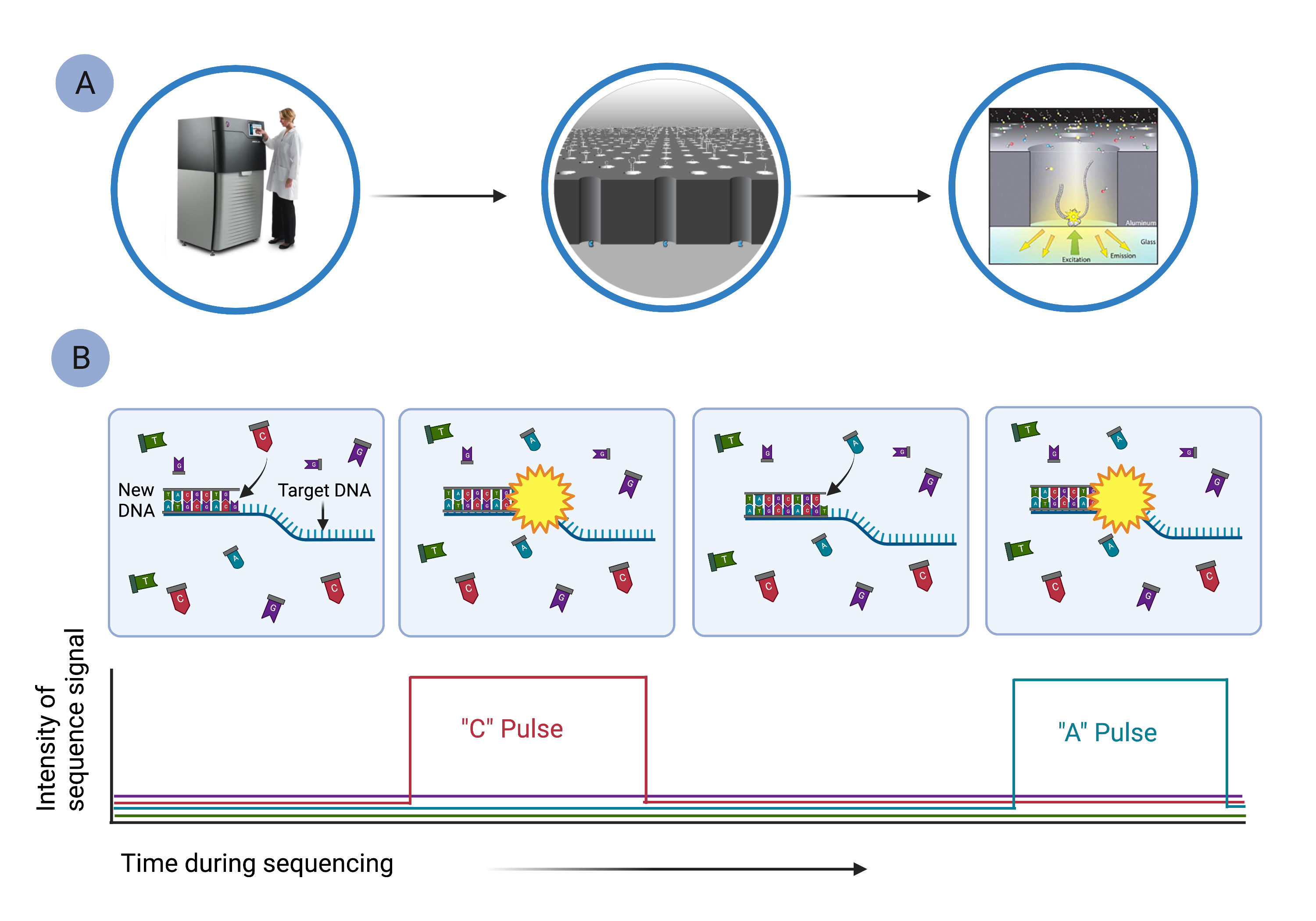
Figure 9. Pacific Biosciences’ single molecule, real-time sequencing can sequence long segments of DNA. A) Long segments of DNA are diluted into microscopic wells so that only a single template strand of DNA is in any well. B) The target DNA segment is used as a template to synthesize a new strand of DNA, using fluorescently labeled nucleotides. As labeled bases are incorporated, they emit a characteristic fluorescent signal that is detected by a camera imaging each cell simultaneously. The complete sequence of the original DNA region is reconstructed using computer software.
The long-read sequencing platform was initially developed by Oxford Nanopore Technologies and does not use a sequencing-by-synthesis approach. Instead, long DNA fragments are pushed through tiny molecular pores (nanopores) in the surface of a special membrane (Fig. 10A).[11] An electrical current is applied across the surface of the membrane, and as the DNA is drawn through the nanopore, it causes a fluctuation in the electrical current (Fig. 10B). The sequencer determines the identity of the nucleotides passing through the pore from the distinct changes in electrical current (Fig. 10B).
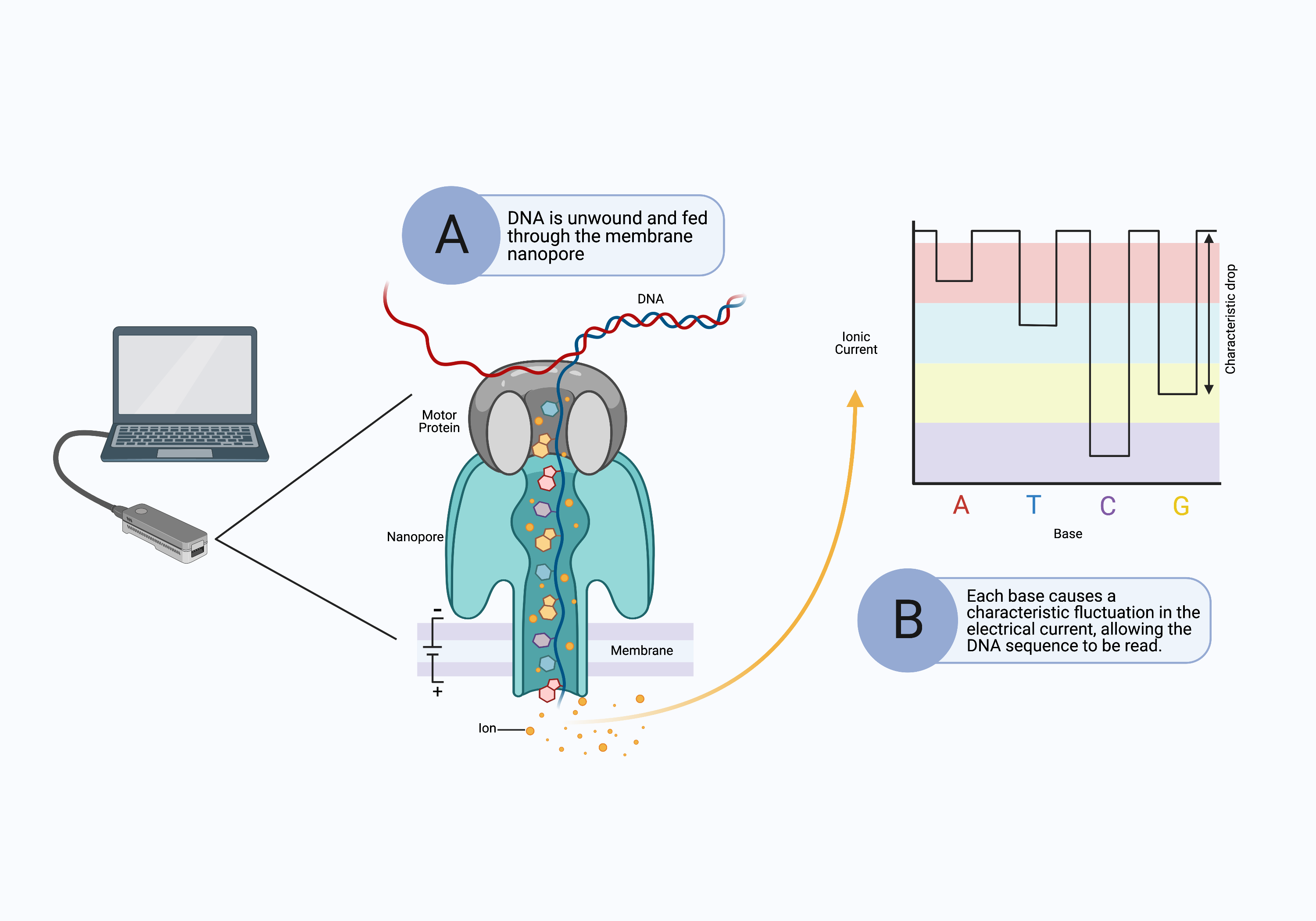
Figure 10. Nanopore sequencing performs long-read sequencing without synthesizing complementary DNA strands. A) A long strand of DNA is unraveled and pushed through a nanopore in a membrane. The motor protein ensures the DNA passes through the nanopore slowly enough to be read. B) As the DNA bases pass through the nanopore, they cause characteristic fluctuations in the electrical current across the membrane that identify the bases. Computer software reconstructs the full sequence of the original DNA segment.
The nanopore approach to sequencing does not require a large volume of additional chemical reagents, unlike those based on sequencing-by-synthesis. The technology is therefore easily miniaturized into portable devices no bigger than a cellphone. Portable sequencing devices have been used in many cases of genomic surveillance, like tracking the spread and evolution of a virus during an epidemic or monitoring the presence of animal populations through trace levels of DNA discarded in the environment. Since its invention, the capillary-based nanopore has been integrated into technologies developed by other companies.
Common Sources of Error and Determining Which Technology to Use
All sequencing technologies are limited by the quality and quantity of DNA used for sequencing. DNA that is old, degraded, or exposed to chemical contaminants can result in numerous errors during sequencing. Similarly, DNA samples that include contaminating DNA from other individuals or other organisms (e.g., bacteria, wildlife) can produce inaccurate sequencing results. Very small amounts of the DNA itself can also cause errors during sequencing, producing a low level of signal during the sequencing process.
DNA quality and quantity limits differ between sequencing technologies and methods, but these limits are often specified by manufacturers and method developers of the sequencing technology. Assessing the quality and quantity of DNA used for sequencing is an important step to gauge the quality and reliability of the sequencing results.
The most common errors that occur in sequencing are random technical errors, such as the incorrect nucleotide base being incorporated at a specific site or the computer failing to differentiate between the fluorescent signals of incorporated bases. A common approach to overcome these errors is to attain a high level of coverage by sequencing the same site multiple times over, providing a consensus for each site. In most cases, coverage of 30x–50x (each base is read 30–50 times on average) is sufficient for high-quality sequencing. Higher coverage may be used when more accuracy is required, for example sequencing a few cancer genes in a patient may use 100x–300x coverage. However, higher coverage comes at a higher financial cost.
Deciding which sequencing technology to use depends on the lab’s familiarity with the sequencing technology (i.e., level of internal validation) and on the specific purpose. For example, the speed and accuracy of second-generation sequencing has made it a common technology in research and medical genetic testing, although nanopore technologies are quickly becoming more popular. In situations that require more mobility, like rapid DNA testing for illegal fishing while out at sea, a small and portable nanopore sequencing device may be preferred.
Technologies may also be used together to complement each other, such as when Sanger sequencing is used to validate medical genetics test results from either short- or long-read sequencers. Collecting complementary sequencing results from multiple different technologies may require coordination between multiple labs. This can introduce potential errors during the transfer of DNA samples (e.g., swapped labels, contamination, degradation). The levels and consistency of internal validation may also vary across labs. These are details that can be reviewed when evaluating DNA sequencing evidence from multiple labs and technologies.
[1] Michael L. Metzker, Emerging Technologies in DNA Sequencing, 15 Genome Resch. 1767 (2005), available at https://doi.org/10.1101/gr.3770505; Jay Shendure & Hanlee Ji, Next-Generation DNA Sequencing, 26 Nature Biotech. 1135 (2008), available at https://doi.org/10.1038/nbt1486.
[2] Vikas Bansal & Christina Boucher, Sequencing Technologies and Analyses: Where Have We Been and Where Are We Going?, 18 iScience 37 (2019), available at https://doi.org/10.1016/j.isci.2019.06.035; Shendure & Ji, supra note 1;Shendure & Ji, supra note 1; Vivien Marx, Method of the Year: Long-Read Sequencing, 20, Nature Methods 6 (2023), available at https://www.nature.com/articles/s41592-022-01730-w.
[3] Adapted from Bansal & Boucher, supra note 2.
[4] Shawn E. Levy & Richard M. Myers, Advancements in Next-Generation Sequencing, 17 Ann. Rev. of Genomics & Hum. Genetics, Aug. 2016, at 95–115, available at https://doi.org/10.1146/annurev-genom-083115-022413; Michael L. Metzker, Sequencing Technologies—The Next Generation, 11 Nature Reviews Genetics 31 (2010), available at https://doi.org/10.1038/nrg2626; Shendure & Ji, supra note 1.
[5] Levy & Myers, supra note 4; Metzker, supra note 4.
[6] Bansal & Boucher, Marx supra note 2.
[7] Søren M. Karst et al., High-Accuracy Long-Read Amplicon Sequences Using Unique Molecular Identifiers with Nanopore or PacBio Sequencing, 18 Nature Methods 165 (2021), available at https://doi.org/10.1038/s41592-020-01041-y; Aaron M. Wenger et al., Accurate Circular Consensus Long-Read Sequencing Improves Variant Detection and Assembly of a Human Genome, 37 Nature Biotech. 1155 (2019), available at https://doi.org/10.1038/s41587-019-0217-9.
[8] Levy & Myers, supra note 4.
[9] Anthony Rhoads & Kin Fai Au, PacBio Sequencing and Its Applications, 13 Genomics, Proteomics & Bioinformatics 278 (2015), available at https://doi.org/10.1016/j.gpb.2015.08.002.
[10] Jin H.Bae et al., Single Duplex DNA Sequencing with CODEC Detects Mutations with High Sensitivity, 55 Nature Genetics 187 (2023), available at https://doi.org/10.1038/s41588-023-01376-0.
[11] Hengyun Lu et al., Oxford Nanopore MinION Sequencing and Genome Assembly, 14 Genomics, Proteomics & Bioinformatics 265 (2015), available at https://doi.org/10.1016/j.gpb.2016.05.004.